
Applications
 Part of the Oxford Instruments Group
Part of the Oxford Instruments Group
Expand
Collapse
Part of the Oxford Instruments Group
Case Studies
Author: Dr. Coralie Spiegelhalter and colleagues,Institut de Génétique et de Biologie Moléculaire and other
Published: 10 Feb 2021 · Last updated: 10 Feb 2021
Cell Biology
Researchers at the Institut de Génétique et de Biologie Moléculaire et Cellulaire in Illkirch, France have developed a new method for correlating live-cell imaging with electron microscopy (EM) on cryofixed samples. According to Yannick Schwab, one of the research team’s members, the group’s goal was to develop a way to correlate light microscopy data acquired from living cells with ultrastructural analysis of the same objects.
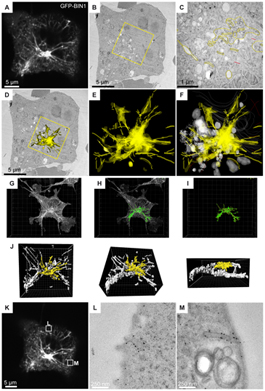
Figure: COS-1 cells transfected with GFP-tagged BIN1 were imaged by confocal microscopy, fixed by high pressure freezing, processed for 60 nm thick serial sections, and labeled with anti-GFP antibody and gold particles. (A) Representative image (z projection of 3 confocal sections) of the fluorescent B1N1 tubules radiating from/to the perinuclear region. (B) One serial thin section showing the transmission-EM image of the same cell. (C) At higher magnification, the immunostaining is visible over various perinuclear structures (highlighted in yellow). Unstained organelles such as mitochondria, Golgi complex, and endosomal vesicles are shown in white. (D) Rendering the entire stack of serial sections allows the immunogold labeling to be projected onto the representative transmission-EM picture, showing a good correlation with the dense perinuclear GFP signal recorded on living cells. (E and F) The distribution of the anti GFP immunogold staining in the volume of the perinuclear region. (G–J) The full z-stack recording was processed in Imaris to reconstruct a 3D model (green) of the GFP fluorescence. The region analyzed by EM (yellow) shows a good correlation with the gold labeling in D-F. (K–M) Correlation analysis of other z planes showed localization of GFP-BIN1 at plasma membrane invaginations. Image reprinted from PLoS One.
To accomplish this, the researchers pre-patterned a reference grid on aclar discs, which their cells were then cultured on. These discs are compatible with cryofixation and resin embedding. Moreover, the co-ordinates on the grid are conserved throughout preparation and EM imaging. The advantage of using this method is that it is compatible with various EM techniques, high-resolution live recording of dynamic phenomena, and fast high-pressure freezing.
In the initial phase of the experiment, the group acquired live-cell imaging data using either a confocal or epifluorescence microscope. Following this, the cells were subjected to either chemical fixation for scanning EM or high pressure freezing for electron tomography (or serial immuno-EM). Lastly, the data acquired from both microscope modalities were arranged to be analyzed and correlated in the Imaris 3D/4D image analysis software.
Using a specific 3D intensity thresholding function, unique to the Imaris software, the researchers were able to select specific objects in the data based on their intensity profiles. Once all of the objects in the data set were identified, Schwab’s group filtered away all of the smaller, unwanted objects in order to isolate only the structures of interest. In combination, the specific thresholding and 3D filtering applications in Imaris allowed the precise determination of the shape and location of specific membrane tubules that form in the perinuclear region of cells transfected with GFP-tagged BIN1.
In the next phase of the experiment, the data extracted from the analysis in Imaris was used to select a set of thin sections to study at the EM level. Using these sections, the researchers tried to elucidate an immuno-EM correlation. From this final analysis, Schwab’s group ascertained that the anti-GFP immunogold distribution across several serial EM sections matched the fluorescent pattern highlighted in Imaris (see figure)
“We managed to get ultrastructural snapshots of dynamic events or to study the 3-D organization of selected subcellular compartments, both with confocal microscopy and with serial or electron tomography,” Schwab said.
Research Paper: Spiegelhalter C, Tosch V, Hentsch D, Koch M, Kessler P, et al. 2010 From Dynamic Live Cell Imaging to 3D Ultrastructure: Novel Integrated Methods for High Pressure Freezing and Correlative Light-Electron Microscopy. PLoS ONE 5(2): e9014. doi:10.1371/journal.pone.0009014.

© Oxford Instruments 2026
